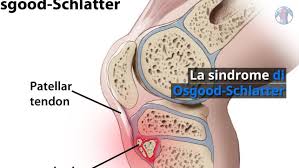
El síndrome de Osgood-Schlatter (OSS) es la causa más común
de dolor de rodilla en adolescentes, especialmente en aquellos que participan en deportes.
ETIOLOGÍA
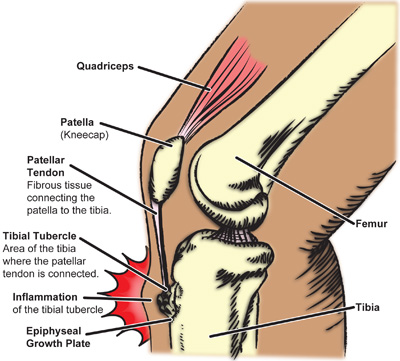
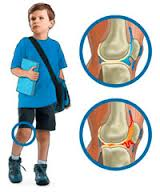
Se considera que la etiología de la enfermedad de Osgood-Schlatter es traumática por tracción excesiva del tendón rotuliano sobre su inserción epifisaria inmadura, lo que provoca microfracturas por avulsión.
Otra causa reportada ha sido la falta de crecimiento de los cuadriceps en comparación con el fémur. Durante un crecimiento repentino en un niño, el alargamiento del músculo es incapaz de mantenerse al día con el fémur que se alarga rápidamente, lo que resulta en un aumento de la fuerza de tracción en la tuberosidad tibial.
INCIDENCIA
La incidencia se ha informado en un 21% entre los atletas adolescentes en comparación con el 4,5% entre los adolescentes no deportistas. Sin embargo, no es específico del atleta. Entre la población general de adolescentes, la tasa de incidencia ha sido reportada en 9.8%
SIGNOS Y SÍNTOMAS
- Se presenta en niños y niñas en crecimiento como dolor local, hinchazón y sensibilidad sobre la tuberosidad tibial en la inserción del tendón patelar. La apófisis puede agrandarse en etapas posteriores.
- El dolor se provocará durante el ejercicio (p. Ej., Correr, saltar) o con contacto directo, como al arrodillarse.
- El dolor se asocia con una extensión de rodilla resistida.
- El paciente puede informar síntomas bilaterales, que están presentes en 20 a 30% de los casos.
- El inicio de los síntomas es gradual.
FACTORES DE RIESGO
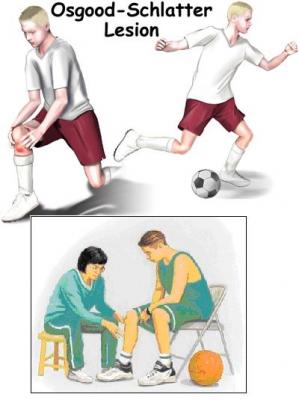
- Niños preadolescentes y adolescentes, generalmente al comienzo de un crecimiento acelerado
- Niños: edades 11-15 años.
- Niñas: de 8 a 13 años.
- Más común en niños.
- Actividades relacionadas con la extensión repetitiva y forzada de la rodilla (contracciones del cuádriceps), como saltar o patear.Aquellos que participan en deportes como el fútbol americano, voleibol, baloncesto, fútbol, gimnasia y patinaje artístico son más susceptibles a una mayor carga del tendón rotuliano y tienen un mayor riesgo de desarrollarlo.
- El aumento del tiempo de participación en el entrenamiento deportivo específico durante la infancia
y la adolescencia aumenta el riesgo de desarrollar OSS y otras lesiones por sobreuso.
DIAGNÓSTICO
- La radiografía es la opción de imagen inicial
- La resonancia magnética (IRM) debe ordenarse cuando el dolor de rodilla es persistente, las radiografías iniciales no son diagnósticas (radiografías normales o derrame articular) y los síntomas de la rodilla indican un trastorno interno.
MANEJO MEDICO
- Las inyecciones de corticosteroides no se recomiendan debido a los informes de casos de atrofia subcutánea.
- Las inyecciones con lidocaína y la adición de dextrosa se asocian con una mejoría en los síntomas y la participación en los deportes en comparación con la lidocaína sola o el cuidado conservador típico para los atletas infantiles con OSS crónico.
TRATAMIENTO FISIOTERAPEUTICO
- Ejercicios terapéuticos y educación del paciente como componentes principales.
- Uso del estiramiento de la musculatura estrecha dentro del rango sin dolor para evitar el riesgo de causar una fractura por avulsión del tubérculo tibial
- Fortalecimiento apropiado, principalmente los cuadriceps. Los ejercicios deben realizarse solo si no presentan dolor para disminuir el riesgo de fractura por avulsión del tubérculo tibial. Puede ser necesario comenzar con ejercicios isométricos de cuádriceps si el paciente presenta atrofia y dolor significativo.
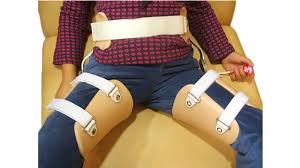
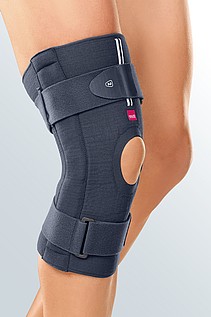
- Ortesis de rodilla para administrar la OSS, pero muchos expertos están de acuerdo en que el uso de la ortesis de rodilla puede ser beneficioso en el manejo de los síntomas. Las ortesis más comunes diseñadas para pacientes con OSS son el contrafuerte de media luna infrapatelar y la ortesis de rodilla patelofemoral con contrafuerte H.
Bibliografías:
OSGOOD SCHLATER Por: Michael Granado, PT, MPT, ATC, CSCS, Cinahl Information Systems, Glendale, CA Andrea Callanen, MPT, Cinahl Information Systems, Glendale, CA
Editado por: Sharon Richman, MSPT, Cinahl Information Systems, Glendale, CA