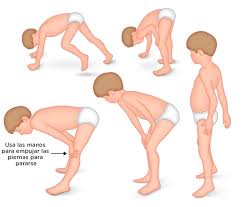
Tipo de epilepsia catastrófica de primer año de vida, caracterizada por espasmos clínicos, retraso del desarrollo psicomotor y patón electroencefalográfico de hipsarritmia
Incidencia
*De 2 a 5 /10000 recién nacidos
*90% de los afectos inician el primer año de vida
*Síntomas entre 3 y 7 meses
*Levemente predomina en hombres
Fisiopatología
Falta de equilibrio entre los neurotransmisores del tallo cerebral
Etiología
Sintomático: Retraso en el desarrollo en el inicio de los espasmos
Criptogénico: No se logra identificar etiología precipitante
Las causas más comunes de espasmos infantiles son:
*Encefalopatía hipóxicoisquémica 10%
*Anomalías cromosómicas 8%
*Malformaciones 8%
*Accidente cerebrovascular perinatal 8%
*Complejo de esclerosis tuberosa 7%
*Hemorragia cerebral 5%
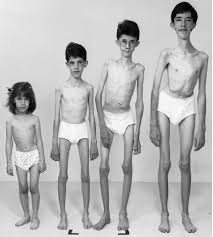
Cuadro clinico
*Espasmos epilépticos
*Retraso del desarrollo psicomotor
*Patón electroencefalogáfico de hipsarritmia
Diagnóstico
*EEG
*Neuroimagen
*Resonancia magnética
*Pruebas genéticas
Tratamiento medico
Fármacos para el control de este padecimiento como ACTH, prednisolona, hidrocortisona, vigabatrina, nitrazepam y ácido valproico
Tratamiento fisioterapéutico
Metodos de neurofacilitación:
*Bobath
*Kabath
*Rood
Pronóstico
Depende de las causas y del momento que inicie el tratamiento
*75-90% de los pacientes presentan regreso metal
*50-60% tienen convulsiones recurrentes a los 5 años de edad
*27-50% desarrollan síndrome de Lennox-Gastaut
Bibliografías:
Sanz-Arrazola, H., & Andia-Berazain, C. (2014). Síndrome de West: Etiología, fisiopatología, aspectos clínicos, diagnóstico, tratamiento y pronostico. Revista Médico-Científica» Luz y Vida», 5(1), 30-35.
La distrodia de Becker es un trastorno hereditario que consiste en una debilidad muscular de las piernas y de la pelvis que empeora lentamente. Es causada por mutaciones en el gen DMD, se hereda de forma recesiva ligada al cromosoma X en los varones Nula producción de distrofina.
INCIDENCIA: Varía entre 1 por cada 18 mil y 1 por cada 31.000 nacimientos en varones
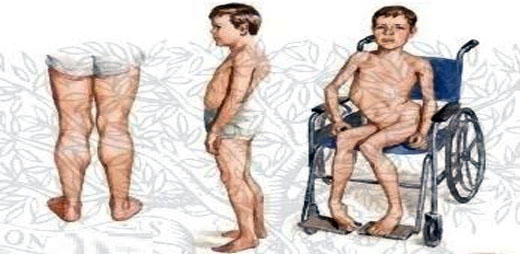
SÍNTOMAS
Contracturas del tendón de Aquiles y bandas iliotibiales, posturas lordoticas, problemas cognitivos, problemas cardíacos y dificultad para deglutir o respirar.
Debilidad muscular de cadera, pelvis, miembros inferiores la pérdida de tejido muscular no es dolorosa, progresión a debilidad proximal y flexores de cuello, escoliosis restrictiva. Torpeza, caídas frecuentes, marcha de pato y dificultad para subir las escaleras.
Aparecen más tarde entre los 5 y 15 años de edad Sobreviven hasta los 40 años de edad
TRATAMIENTO
No existe cura. El tratamiento tiene como objetivo controlar los síntomas y mejor la calidad de vida por medio de la terapia física. En terapia física el uso de dispositivos de ayuda para estimular los músculos tensos, terapia ocupacional ayuda a mejorar las habilidades AVD.
La disfrofia muscular de Duchenne esun trastorno de origen genético caracterizado por la debilidad muscular progresiva que ocurre debido a falta de distrofina, una proteína fabricada por las fibras musculares. La falta de un fragmento o la modificación del gen de la distrofina hacen que la persona carezca de esta proteína. Gen responsable es recesivo y ligado al cromosoma X. INCIDENCIA 1 por cada 3.300 nacimientos en varones SÍNTOMAS Comúnmente aparecen antes de los 6 años de edad, pueden darse incluso en el periodo de lactancia: fatiga, discapacidad intelectual, problemas de aprendizaje, dificultad para respirar, dificultad para levantarse de una posición Existe una Debilidad muscular que es generalizada, pero comienza en piernas y pelvis, hay menos gravedad en brazos y cuello, caídas frecuentes, dificultad para cambiar de posición y un empeoramiento gradual de la debilidad muscular.
PRONOSTICO Suele pasar desapercibida hasta los 3-5 años de edad Esperanza de vida no supera los 30 años TRATAMIENTO No existe una cura conocida para la distrofia. El objetivo del tratamiento es controlar los síntomas para optimizar la calidad de vida. Los esteroides pueden disminuir la pérdida de fuerza muscular PRUEBAS Y EXAMENES Concentración de creatina cinasa (CCK) Biopsia muscular Pruebas genéticas.
BIBLIOGRAFÍAS Viñet Espinosa L.M. (2018). Distrofia muscular de Duchenne. A propósito de un caso. Panorama. Cuba y Salud. Vol 13.
Quesada Vargas M., Esquivel Rodríguez N., Rosales Guitiérrez J.M. (2019). Distrofia muscular de Duchenne: diagnóstico y tratamiento. Revista Médica Sinergia. Vol. 4 Num 12.
La espina bífida es una afección que afecta la columna vertebral y suele ser evidente en el nacimiento. Es un tipo de defecto del tubo neural (DTN).
La espina bífida puede aparecer en cualquier lugar a lo largo de la columna si el tubo neural no se cierra por completo. La columna vertebral que protege la médula espinal no se forma y no se cierra como debería. Eso suele producir daño de la médula espinal y los nervios.
La espina bífida puede provocar discapacidades físicas e intelectuales, que van de leves a graves. La gravedad depende de lo siguiente:
El tamaño y la localización de la abertura en la columna.
Si parte de la médula espinal y los nervios están afectados.
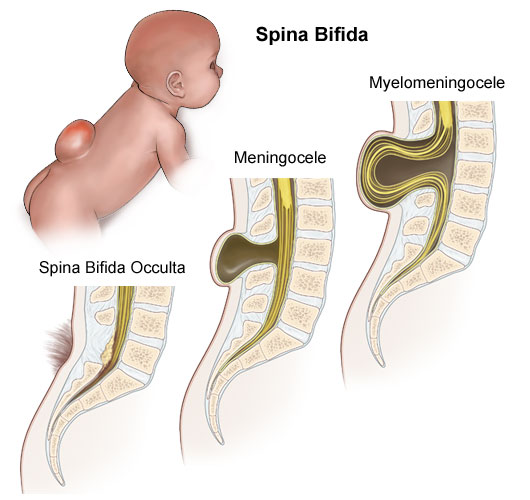
Tipos de espina bífida
Los tres tipos más comunes de espina bífida son los siguientes:
Mielomeningocele
Cuando se habla de espina bífida, la mayoría de las veces se habla de mielomeningocele. El mielomeningocele es el tipo más grave de espina bífida. Con esta afección, un saco de líquido sale a través de una abertura en la espalda del bebé. Parte de la médula espinal y los nervios están en ese saco y presentan daños. Ese tipo de espina bífida provoca discapacidades que pueden ser de moderadas a graves, como problemas que afectan la forma en que se va al baño, pérdida de sensibilidad en las piernas o los pies, o no poder mover las piernas.
Meningocele
Otro tipo de espina bífida es el meningocele. Con el meningocele, un saco de líquido sale a través de una abertura en la espalda del bebé. Pero la médula espinal no está en ese saco. Por lo general, el daño de los nervios es escaso o nulo. Este tipo de espina bífida puede provocar discapacidades menores.
Spina Bifida Occulta
La espina bífida oculta es el tipo más leve de espina bífida. En este caso, la espina bífida está “escondida”. En esta enfermedad, hay un pequeño hueco en la columna, pero no hay una abertura ni un saco en la espalda. La médula espinal y los nervios suelen ser normales. Muchas veces, la espina bífida oculta recién se detecta en la niñez avanzada o la adultez. Ese tipo de espina bífida no suele provocar discapacidades.
Diagnóstico
La espina bífida se puede diagnosticar durante el embarazo o tras el nacimiento del bebé. La espina bífida oculta puede no ser diagnosticada hasta finales de la infancia o la edad adulta, o tal vez nunca ser diagnosticada.
Durante el embarazo
Durante el embarazo, hay pruebas de detección (pruebas prenatales) que se usan para determinar si el bebé tiene espina bífida u otros defectos congénitos. Hable con su médico si tiene alguna pregunta o inquietud sobre estas pruebas prenatales.
AFP: AFP quiere decir alfafetoproteína, una proteína que produce el bebé en gestación. Es un simple análisis de sangre que mide qué nivel de AFP pasó del bebé a la sangre de la madre. Un nivel alto de AFP podría indicar que el bebé tiene espina bífida. Un análisis de AFP puede formar parte de una prueba llamada “de triple detección”, que detecta defectos del tubo neural y otros problemas.
Ecografía: una ecografía es un tipo de imagen que se toma del bebé. En algunos casos, el médico puede ver si el bebé tiene espina bífida o hallar otros motivos por los que podría haber un nivel alto de AFP. Con frecuencia, la espina bífida se puede ver con esta prueba.
Amniocentesis: en esta prueba, el médico toma una pequeña muestra del líquido amniótico que rodea al bebé en el útero. Un nivel de AFP más alto que el promedio en el líquido podría indicar que el bebé tiene espina bífida.
Después del nacimiento del bebé
En algunos casos, es posible que no se diagnostique la espina bífida hasta después del nacimiento del bebé.
A veces hay una zona de piel con pelo o un hoyuelo en la espalda del bebé que se detecta por primera vez después del nacimiento. El médico puede usar estudios por imágenes, como radiografías, resonancias magnéticas o tomografías computadas, para ver la columna y los huesos de la espalda del bebé con mayor claridad.
A veces, la espina bífida recién se diagnostica después del nacimiento del bebé porque la madre no recibió atención prenatal o porque una ecografía no mostró imágenes claras de la parte afectada de la columna.
Tratamientos
No todas las personas que nacen con espina bífida tienen las mismas necesidades, así que el tratamiento es diferente para cada persona. Algunas personas tienen problemas más graves que otras. Las personas con mielomeningocele y meningocele necesitan más tratamientos que las que tienen espina bífida oculta.
Causas y prevención
No se conocen todas las causas de la espina bífida. Es necesario estudiar más el rol de factores como los genes y el medio ambiente en la aparición de la espina bífida. Sin embargo, sabemos que hay maneras en que las mujeres pueden ayudar, antes y durante el embarazo, a reducir el riesgo de tener un bebé con espina bífida.
Si está embarazada o podría quedar embarazada, siga estos consejos para ayudar a evitar que su bebé tenga espina bífida:
Tome 400 microgramos (mcg) de ácido fólico todos los días. Si ya ha tenido un embarazo afectado por espina bífida, es posible que tenga que tomar una dosis más alta de ácido fólico antes de quedar embarazada y durante los primeros meses de embarazo. Hable con su médico para analizar qué es lo mejor para usted.
Hable con su médico o farmacéutico sobre todos los fármacos recetados y de venta libre, vitaminas y suplementos dietarios o a base de hierbas que esté tomando. Si tiene una afección médica, como diabetes u obesidad, asegúrese de que el problema esté controlado antes de quedar embarazada.
Evite calentar demasiado su cuerpo, por ejemplo, en un jacuzzi o sauna.
Siempre que tenga fiebre, trátela de inmediato con Tylenol (u otra marca de acetaminofeno).
Recuerde:
La espina bífida aparece en las primeras semanas de embarazo, a menudo antes de que la mujer sepa que está embarazada. Aunque usar ácido fólico no es una garantía de que la mujer tendrá un embarazo sano, tomar ácido fólico puede ayudar a reducir el riesgo de que una mujer tenga un bebé con espina bífida. Dado que la mitad de todos los embarazos en los Estados Unidos no se planifican, es importante que todas las mujeres que pueden quedar embarazadas tomen 400 mcg de ácido fólico al día antes de quedar embarazadas y durante los primeros meses de embarazo.
REFERENCIAS:
Russel F, Najat F, El kahabt, et al. Shun complication in children with mielomeningocele: effect of timming of shunt placement clinical article J. neurosuregy 2009 jun 3: 526-522.
Díaz Sanhueza, C., Pardo Vargas, R. A., & Bustos, P. (2018). Manifestaciones neurológicas asociadas a espina bífida en niños. Medicina de Familia. SEMERGEN, 44(4), 276-280.
Es una enfermedad infecciosa de origen viral que infecta a niños entre 2-6 años. En el año 1988 la OMS crea vacuna. En México esta enfermedad se encuentra erradicada.
El poliovirus va de la boca y se multiplica en el intestino se transmite de una persona a otra a través de la materia fecal, de igual forma este puede propagarse a través de alimentos contaminados, su incubación consiste en un periodo desde el contacto hasta los síntomas y signos clínicos de 8 a 12 días. Posterior a uno años se produce el Sindrome postpoliomelitis
En la poliomelitis subclínia el paciente no advierte ningún síntoma Poliomelitis paralítica los síntomas son muy graves pueden ocasionar la muerte Poliomelitis no paralítica presentan síntomas leves tos y gripe duran 72 hrs hay dolor de cabeza, dolor y enrojecimiento de garganta. fiebre leve, vómitos, fatiga, reflejos anormales, dolor o rigidez en cuello que impide inclinar hacía delante. Poliomelitis no paralítica presentan síntomas leves tos y gripe duran 72 hrs hay dolor de cabeza, dolor y enrojecimiento de garganta. fiebre leve, vómitos, fatiga, reflejos anormales, dolor o rigidez en cuello que impide inclinar hacía delante. Poliomelitis paralítica se presentan síntomas de la no paralitica además de: Pérdida de reflejos Dolor y espasmos musculares intensos Debilidad y flacidez en las extremidades a veces perceptible en un solo lado del cuerpo Parálisis repentina (temporal o permanente) Deformidad en extremidades
PREVENCIÓN La vacuna es el único método eficaz para prevenir la poliomelitis y existen dos tipos. SABIN: Virus vivos atenuados (VPO) SALRK: Virus muertos o inactivados (VPI) DIAGNOSTICO Aislamiento 7 o 14 días después de la aparición TRATAMIENTO Descanso, analgésicos, antibióticos, respiradores portatiles. PACIENTES CON POSTPOLIO Disminuir dolor Disminuir espasmos o contracturas Fisioterapia respiratoria Evitar deformidades Alineación de segmentos Fuerza muscular e inestabilidad articular
REFERENCIAS
Russell WR, Fischer-Williams M: Recovery of muscular strength after poliomyelitis. Lancet 1954, 266:330–333.
Cooksey FS: The role of physiotherapy in the treatment of poliomyelitis. Proc Royal Soc Med 1948, 41(6):395–401.
WHO: Vaccine Preventable Diseases. 2011. Available at http://www.who.int/ immunization/monitoring_surveillance/burden/vpd/surveillance_type/ active/poliomyelitis_standards/en/.
Little Club Oxford: la PC es una alteración del tono, postura y movimiento.
Bobath: la PC es un grupo de condiciones resultantes del daño o mal desarrollo del cerebro. la lesión es estacionaria.
La PC es causada cuando las células del cerebro que son responsables control muscular y movimiento fallan en tener un adecuado desarrollo o mueren como resultado de la lesión.
EPIDEMIOLOGÍA: 2-3 casos por cada 1000 recién nacidos, la proporción de hombres a mujeres es de 1.3-1. Mayor prevalencia en niños prematuros.
FISIOPATOLÓGIA: alteraciones motoras que resultan del efecto anatómico y electrofisiológica sobre neuronas somáticas y autónomas localizadas en la corteza motora, ganglios basales y cerebelo.
POSIBLES CAUSAS
Migración de células cerebrales: en el feto en desarrollo las células se diferencian y mueren por formar un tipo de célula apropiada se interrumpe el proceso. Un gran número de condiciones neurológicas se pueden desarrollar.
Mielinización: pobre mielinización prenatal de células nerviosas. Las neuronas no están protegidas y se dañan.
Muerte celular: durante el parto muchas células pueden morir como resultado de asfixia o pérdida de sangre. Más común en partos urgentes como cesárea, sufrimiento fetal.
Sinapsis: pueden generarse en postpartos. Puede lesionar el cerebro y producir PC, se incluyen infecciones, traumas y asfixia.
FACTORES DE RIESGO
Ambiental: infecciones, malformaciones (antes, durante, después del parto).
SEVERA: dependencia, déficit de comunicación, depende de material adaptado.
PLEJÍA: ausencia de movimiento voluntario.
PARESIA: ausencia parcial de movimiento voluntario.
TONO MUSCULAR
Espástica: más frecuente 70-80%
Discinética: menos frecuente 10-15%
Atáxica: mas infrecuente.
ESPÁSTICA: afectación de la corteza motora/ vías subcorticales intracerebrales principalmente de la vía piramidal. Hay hipertonía puede ser espasticidad o rigidez.
DISCINÉTICA: lesión de la vía extrapiramidal y ganglios basales(núcleos de base y conexiones) caudado, putamen, pálido o subtalámico. Alteraciones de tono con fluctuaciones y cambios bruscos del mismo. Movimientos involuntarios y reflejos arcaicos.
ATÁXICA: daño en el cerebelo o sus vías. Hay hipotonía, temblor intencional, disartria.
MIXTA: combinación de diferentes tipos de PC. Trastornos motores/ extrapiramidales. Alteración de tono.
DIAGNÓSTICO LIVINE: estrabismo, alteración del tono, alteración de reflejos, patrones anormales de postura y movimiento, alteración en el inicio y evolución de reacciones posturales. 8debe presentar mínimo 4 signos).
TRATAMIENTO
Quirúrgico: cirugía multinivel/ cirugía de deformidades.
Médico: toxina botulínica, bloqueadores receptivos de receptor de dopamina, aparatos ortopédicos.
EXAMINACIÓN/EVALUACIÓN: datos relacionados con el usuario del paciente y el entorno – ambiente en que se desenvuelve. Datos sobre su salud general y de la razón de consulta.
Anamnesis: edad, genero, etnia, educación.
Historia social: creencias, conductas.
Creencias y desarrollo.
Historia familiar: antecedentes personales, quirúrgicos, traumáticos, medicamentosos.
Estado de salud general.
Motivo de consulta: escribir tal cual.
Hábitos de salud.
Ocupación.
Otras pruebas.
«NO OLVIDAR LA ESPECTATIVA DEL PACIENTE»
EXAMINACIÓN Y EVALUACIÓN POR SISTEMAS
Sistema tegumentario: cicatrización (aspecto, diámetro, coloración, longitud, pigmentación, hidratación) integridad de la piel.
Sistema osteomuscular: características antropométricas (AMA/ROM, fuerza muscular, postura, flexibilidad).
Sistema neuromuscular: integridad refleja, tono, reflejos patológicos.
TEST Y MEDIDAS
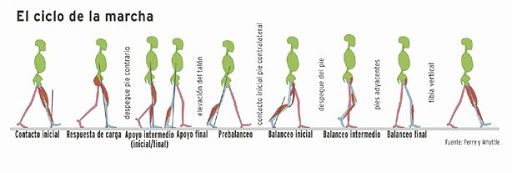
Marcha y balance: análisis de la marcha (balanceo/estático).
Función motora: destreza, coordinación.
Aditamentos y ortesis: dolor, manejo en el hogar, autocuidado e independencia.
Integridad sensorial: test de sensibilidad.
DIAGNÓSTICO: se refiere a la determinación de las capacidades /discapacidades y/o limitaciones funcionales resultantes de enfermedad, lesión, intervención quirúrgica, u otras condiciones de salid, directamente relacionadas con su campo especifico de saber.
PRONÓSTICO: determina las posibilidades de mejoramiento que pueden obtenerse. Establece los resultados esperados en términos de objetivos terapéuticos. Aproxima el tiempo requerido, numero de sesiones e intervalos entre las mismas para el logro de los objetivos.
INTERVENCIÓN: objetivo general, objetivos a corto, mediano y largo plazo (específicos).
RESULTADOS: reevaluación tras el tiempo estipulado en el plan de manejo.
REFERENCIAS Jiménez, E. (2016). Guía metodológica para elaborar el diagnóstico fisioterapéutico según la Clasificación Internacional del Funcionamiento (CIF), de la discapacidad y de la salud. Gac. Med. Bol. 39(1):46-52
Deficiencia: problema en las funciones o estructuras corporales , ya sea una desviación importante o una pérdida.
Limitación: dificultad que puede tener una persona en el desarrollo en la realización de actividades.
Restricción: problemas que un individuo puede experimentar al involucrase en situaciones vitales ya sean sociales o culturales.
Facilitador: es cualquier objeto o sujeto que actúa como instrumento para que las actividades puedan realizarse con menor dificultad o de una manera más sencilla.
Barrera: son todos aquellos impedimentos u obstáculos físicos que limitan la movilidad de los individuos.
CLASIFICACIÓN
Primario: trastorno funcional o estructural que provoca impedimentos secundarios (ataxia, corea, espasticidad).
Secundario: consecuencia del primario o dismovilidad de impedimentos primarios en conjunto (secuela de una lesión primaria).
Estructural: trastorno en la continuidad de una estructura, provocan impedimentos funcionales (fractura).
Funcional: trastorno en función corporal (debilidad muscular, dolor). Fisiológica (hipotonía) y psicológica.
Limitación de la actividad: impedimentos en conjunto que provocan una limitación en actividades de la vida cotidiana.
Restricción de la participación:
AVD básicas: higiene mayor y menor (desplazamiento).
Actividades instrumentales: comprar (uso de locomoción selectiva).
Ocupación. contexto escolar (diversidad de oficios).
Factores ambientales: todos aquellos que se encuentran en el entorno de la persona y que inciden en su discapacidad. barreras arquitectónicas.
Factores personales: son propios de la persona (edad, sexo, etnia, familia etc.)
Factores contextuales: suma de factores ambientales y personales que dan como resultado factores contextuales.
Interacción positiva: condición de salud más factores contextuales = FUNCIONAMIENTO.
Interacción negativa: condición de salud más factores contextuales = DISCAPACIDAD.
REFERENCIA Jiménez, E. (2016). Guía metodológica para elaborar el diagnóstico fisioterapéutico según la Clasificación Internacional del Funcionamiento (CIF), de la discapacidad y de la salud. Gac. Med. Bol. 39(1):46-52
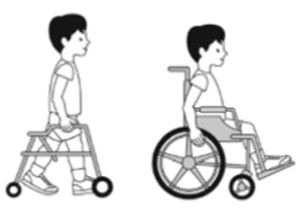
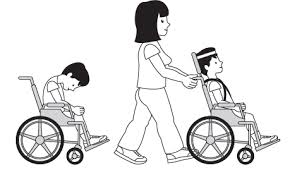
NIVEL III: Anda utilizando un dispositivo con sujeción manual.
NIVEL IV: Autonomia para movilidad con limitación; puede usar sistemas de propulsión a motor.
NIVEL V: Transportado en silla de ruedas manual.
REFERENCIA Palisano, R., Rosenbaum, P., Bartlett, D., Livingston, M. (2007). Clasificación de la Función Motora Gruesa. Dev. Med. Child. Neurol. 39:214-223
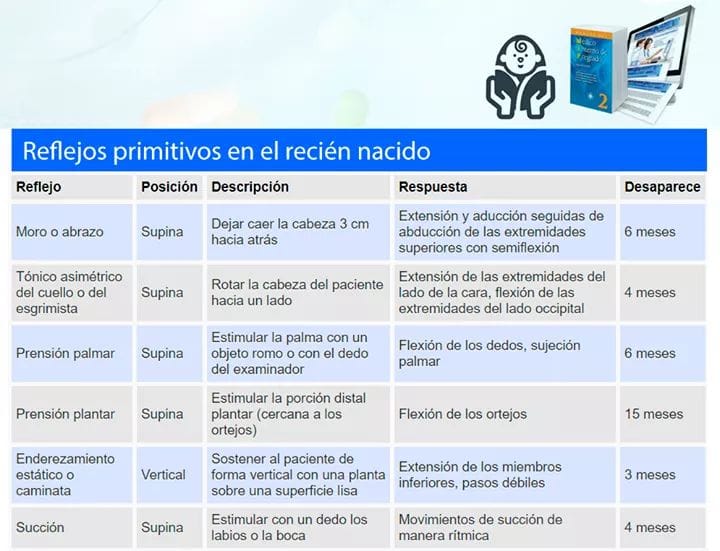
Un reflejo es una reacción muscular involuntaria a cierto tipo de estimulación. Se sabe que ciertas sensaciones o movimientos producen respuestas musculares específicas. La presencia e intensidad de un reflejo es una señal importante de funcionamiento y desarrollo neurológico, y su ausencia es indicativo de que algo no anda bien en el desarrollo del sistema nervioso. Estos reflejos presentes cuando el bebe nace le permiten sobrevivir. Son unos movimientos automáticos dirigidos desde el tronco encefálico, la parte más primitiva de nuestro cerebro y no hay implicación cortical, esto es, no están controlados de forma voluntaria.REFLEJOS DE MESENCEFALO Se encuentran integradas en un nivel más alto en el sistema nervioso central. Enderezamiento cervical Enderezamiento del cuerpo sobre el cuerpo Enderezamiento laberintico Enderezamiento optico Landau
REFERENCIAS O’Sullivans S. Physcal Rehabilitation. Philadelphia. FA. Davis Company. 2001 Gessel A, Matadura C. Educación y Manejo del desarrollo neurofisiológico normal y anormal del niño pequeño y el pre-escolar. Pailos. México 1989. Levitt S. Tratamiento de la parálisis cerebral y retraso psicomotor. Tercera edición. Editorial Panamericana. 2001.
Barinagarrementeria Aldatz F, Dávila Maldonado L, Ló- pez Ruiz M, Marfil Rivera A. Neurología Elemental. 1ª edición. Elsevier. Barcelona España, 2014.